学用BLAST程序进行数据分析
学用BLAST程序进行数据分析
2012/07/13 21:49:32
主要内容
-
基本概念
-
常用BLAST程序介绍
-
BLAST算法简介
-
BLAST常用参数设置
-
本地BLAST的安装步骤
-
本地BLAST的使用
1. 基本概念
相似性(Similarity)
是指序列比对过程中用来描述检测序列和目标序列之间相同或相似碱基或氨基酸残基占全部比对碱基或氨基酸残基的比例的高低,属于量的判断。
同源性(Homology)
是指从某一共同祖先经趋异进化而形成的不同序列。只有当两个蛋白质在进化关系上具有共同的祖先时,才可称它们为同源的,属于质的判断。
相似性和同源性的关系
当相似程度高于50%时,比较容易推测检测序列和目标序列可能是同源序列;
而当相似性程度低于20%时,就难以确定或者根本无法确定其是否具有同源性。
总之不能把相似性和同源性混为一谈。所谓“具有50%同源性”,或“这些序列高度同源”等说法,都是不确切的,应避免使用。
序列相似性比较和同源性分析
序列相似性分析: 就是用来计算待研究序列与某序列之间的相似性程度,常用的软件包有BLAST、FASTA等;
序列同源性分析: 是将待研究与来自不同物种的序列中进行进化分析,以确定该序列与其它序列间的亲源关系。常用的程序包有Phylip及Mega等进化分析软件;
全局比对与局部比对
全局比对
寻找序列在全长范围内最佳比对。
常用算法如:Needleman-Wunschalgorithm(Needle)
在线程序如:Needle
局部比对
寻找序列在局部区域的最高比对打分。
常用算法如:Smith-Waterman algorithm,blast,fasta等
在线程序如:Water
Needle及Water的在线程序<http://bioweb2.pasteur.fr/alignment/intro-en.html>
也可以本地安装Emboss执行以上程序
局部相似性比对的生物学基础
蛋白质功能位点往往是由较短的序列片段组成的,尽管在序列的其它部位可能有插入、删除等突变,但这些关键的功能部位的序列往往具有相当大的保守性。
而局部比对往往比整体比对对这些功能区段具有更高的灵敏度,因此其结果更具生物学意义。
通过以上两个基本概念我们应明白BLAST属于一种局部比对程序,最终比对出的结果是序列之间的相似性。
BLAST 程序常用的两个评介指标
Score:
使用打分矩阵对匹配的片段进行打分,这是对各对氨基酸残基(或碱基)打分求和的结果,一般来说,匹配片段越长、相似性越高则Score值越大,结果越可信。
E-value:
BLAST程序在搜索空间中可随机找到获得这样高分的序列的可能性(期望值),因此E-value越高,则代表结果越有可能是随机获得的,也就越不可信。搜寻空间大小约略等于查询序列的长度乘以全部database序列长度的总和,再乘以一些系数。
我们在获得一个Blast结果时需要看这两个指标。
如果Blast获得的目标序列的Score值越高并且E-value越低表明结果越可信,反之越不可信。
其它的一些重要关键概念
HSP(HighScoring Pair):
在局部比对时,得分高的匹配序列被称为高分值片段。
LCRs(lowcompositional complexity regions):
低复杂度区域,即这些区域的组成有某些偏好,比如DNA中的简单重复序列。在蛋白质中一些残基过多表现。在进行BLAST比较时,将会把LCRs屏蔽掉,防止它们过高评价匹配的显著性。在核酸中用n,在蛋白质中用X代替。
gi(GenBankIndex)
特定于GenBank数据库中所赋予每一条序列的特定索引数字。
nr(non-redundant database)
非冗余数据库,该库信息多,并且无冗余序列
2、常用BLASTBLAST程序
BLAST(BasicLocal Alignment Search Tool)基于匹配短序列片段,并用一种强有力的统计模型来确定未知序列与数据库序列的最佳局部联配的一种程序。
主要的 BLASTBLAST程序
| 程序名 | 查询序列 | 数据库 | 搜索方法 |
| ———- | ———— | ———- | ———— |
| Blastn | 核酸 | 核酸 | 逐一搜索核酸数据库中的序列 |
| Blastp | 蛋白质 | 蛋白质 | 逐一搜索蛋白质数据库中的序列 |
| Blastx | 核酸 | 蛋白质 | 将核酸序列以6种读码框翻译成蛋白质然后和蛋白质数据库中的序列逐一比对。 |
| Tblastn | 蛋白质 | 核酸 | 将蛋白质序列和核酸数据库中的核酸序列6框翻译后的蛋白质序列逐一比对。 |
| TBlastx | 核酸 | 核酸 | 将核酸序列以6种读码框翻译成蛋白质序列再和核酸数据库中的核酸序列以6种读码框翻译成的蛋白质序列逐一进行比对。 |
PSI PSI-BLAST( 位置相关的迭代BLAST)
这个程序主要用来搜索蛋白质的“远亲”。
首先,用户提交的蛋白质序列的所有“近亲”的列表被建立起来,然后这些蛋白质被结合成一种平均的“特征序列”。
再用这个特征序列在蛋白质数据库中进行搜索,就会找出更大的一组蛋白质的列表。再将这个蛋白质列表生成一个不同的特征序列,这个序列被用来迭代地运行上述过程。
通过在搜索中包含相关的蛋白质,PSI-BLAST对于寻找已知蛋白进化上的“远亲”的灵敏度要比一般的blastp高很多。
其它的一些 BLASTBLAST子程序
Gapped BLAST
允许在它产生的比对(alignments)中存在缺口。
Megablast
该程序使用“模糊算法”加快了比较速度,可以用于快速比较两个较长的序列。
discontiguousmegablast
与megablast不同的是主要用来比较来自不同物种之间的相似性较低的分歧序列。
PHI-BLAST
模式发现迭代BLAST。
Bl2seq
给定两个序列,相互进行BLAST比对,快速检查两个序列是否存在相似性片断
Specialized Blast Specialized BLAST pages
CD -Search
是使用RPS -BLAST程序以一个蛋白质序列与保守结构域数据库(Conserved Domain Database) 做比较。
PairwiseBLAST
PairwiseBLAST是用BLAST程序实现两个序列之间的比较。
IgBLAST
IgBLAST被开发出来以便於分析在GenBank中的免疫球蛋白的序列。它允许用blastp或blastn来搜索nr资料库或一个由免疫球蛋白生殖系变化区基因的特殊的资料库。
3、BLASTBLAST算法简介
BLAST 是一种基本局域联配搜索工具,主要用来搜索数据库中相似序列。
它的搜索速度快并且把数据库搜索建立在了严格的统计学基础之上,是目前最常用的同源检索工具,是由AltschulSF et al(1990)提出的一种算法。
BlastBlast的算法流程

BLAST 的基本步骤
将待检索序列分割成长度为w的连续子串
快速找出数据库中所有与固定长度w完全配对的位置
以此位置为起点进行延伸比对,并计算出最高分数
将最高分标准化,并按此分数进行排序
换算成期望值(E-VALUE)
显示出符合Score及E-value的序列
4、BLASTBLAST常用参数设置
在NCBI进行BLAST的操作程序非常简单,只要将你的序列贴进去,点几下鼠标就会得到结果,但是如果能正确的修改一下BLAST的参数,可能你会得到更好的结果!以下我们一起讨论一下如何来修改BLAST的参数!
BLAST 的具体过程:
1.登陆NCBI的BLAST主页 <http://www.ncbi.nlm.nih.gov/BLAST/>
2.根据序列类型及目的选择合适的程序
3.填写表单信息
4.提交任务
5.查看和分析结果
BLAST 程序的选择
在BLAST程序选择上,应尽可能地利用blastp从蛋白质水平进行检索,然后用blastx、tblastn、tblestx从DNA或蛋白质翻译水平进行检索,最后才用blastn进行DNA水平进行检索。
当然如果为非编码序列只有采用blastn进行检索。
E-value 的设置
如果检索的序列较短,可适当的提高E值,否则可能会找不到目的序列,反之如果序列较长可适当提高E值。
通常无论是从DNA水平,还是蛋白质水平进行检索,E值设为1通常可满足要求。
Word size 的选择
BLAST算法将查询序列分割成一系列具有字段长度的小的序列段进行数据库搜索,因此当此值越小得到的搜索结果越多,但假阳性也越多,服务器负担也越重。
对于蛋白质搜索,窗口大小可设置为3或2,默认为3;对于核酸搜索,默认的字段长度是11,可选择7,11或15。 因此如果你对搜索的结果不满意时可以试着降低Word size的值。
打分矩阵的选择
比对所选用的记分矩阵对最终结果影响也很大。
一般高值BLOSUM矩阵和低值PAM矩阵最适合于研究近相关的蛋白质序列。低值BLOSUM矩阵和高值PAM矩阵最适合于研究远相关的蛋白质序列。
一般情况BLOSUM62检测各种蛋白的效果比BLOSUM60和BLOSUM70稍好,比PAM矩阵好得多。
在BLAST五个程序中只有BLASTN不需要这些矩阵,搜索时不必选定。
空位罚分的选择
严紧的罚分很难将本来很相似的序列对准;而松弛的罚分甚至可以使两个无关的序列达到100%的相似性。
一般情况下程序默认的空位罚分(11/1)基本能满足检索要求,但对具体的查询序列,采用不同的空位罚分方法会取得不同的检索效果。
低复杂区域及重复区域的处理
无论是DNA序列类似性检索,还是蛋白质序列类似性检索,一般都应该去除查询序列中的低复杂区域。
就蛋白质序列检索而言,不必去除序列中的重复片段,但对DNA序列检索,就必须去除序列中的重复片段。
5、本地BLASTBLAST的安装
大家一般都做过基于网络的BLAST ,但网络BLAST一般只能搜索一个序列,要搜索多个序列,特别是做大量的数据比较时,网络BLAST几乎是不可能的,这个时候我们就可以考虑做本地BLAST了。
使用本地 BLASTBLAST的原因
1.特殊的数据库要求
2.涉及序列的隐私与价值
3.批量处理
4.与其它本地程序配合使用
5.其他原因??
本地 BLASTBLAST构建步骤
下载BLAST的安装程序
将BLAST保存到适当的位置
点击安装程序来安装BLAST
设置BLAST的相应参数
下载 BLASTBLAST的本地安装程序
可以到NCBI的官方网站下载最新的BLAST程序。下载网址:<ftp://ftp.ncbi.nih.gov/blast/executable>
注意一定要选择和你的计算机操作系统相匹配的程序,如Windows系统要下载“blast-2.2.18-ia32-_win32_.exe”。
本地 BLASTBLAST的相应参数设置
告诉BLAST程序你的数据及数据库放在哪
1.建立一个新的文件并命名为:ncbi.ini
2.在该文件中输入四行数据如下所示:
[NCBI]
Data=“C:\ncbi-blast\data”(你的数据存放的文件夹)
[BLAST]
BLASTDB=“C:\ncbi-blast\db”(数据库存放在的文件夹)
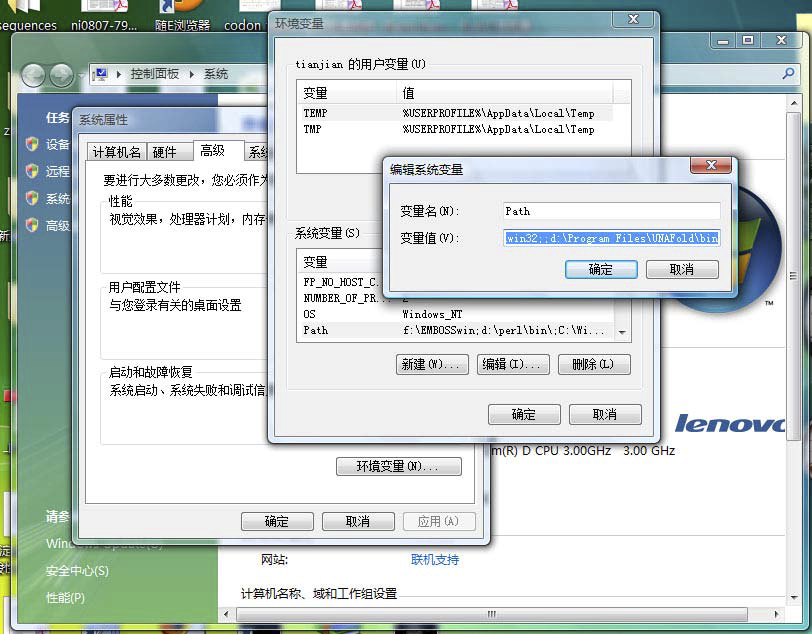
路径设置步骤(系统环境设置-便于命令调用)
右键点击我的电脑
选择属性
再选择系统属性
选择高级标签
选择环境变量
双击path
在路径中填入你的BLAST的可执行文件所在目录
有的时候还需要重新启动电脑
BLASTBLAST的路径设置
6、本地BLASTBLAST的使用
构建本地数据库
进行BLAST搜索
数据库的获取
最简单的方法是直接到NCBI或别的网站去下载
也可以将自己的序列,或与自己工作相关的序列进行整理构建成一个小型的数据库
注意:以上文件格式一般可存为fasta格式
构建 BLASTBLAST用的数据库
将已构建好的数据拷贝到你所设定的数据库所在文件夹
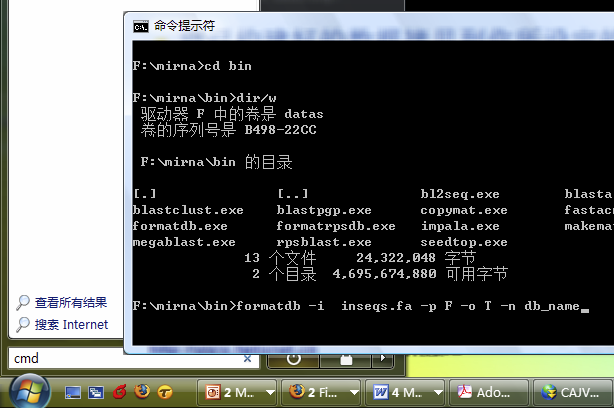
运行cmd命令
在cmd环境中输入如下所示命令
formatdb–i inseqs.fa–p F –o T –n db_name
命令结束后你会发现在你的数据库文件夹里多了一些以db_name开头的文件,这些就是BLAST所需要的一些文件
输入过程
Formatdb 的一些参数说明
-i 输入文件,只能是一个文件
-o Parse options (默认是F) T -True: Parse SeqId and create indexes. F -False: Do not parse SeqId. Do not create indexes
-p 文件类型(默认是T) T -protein F -nucleotide [T/F] Optional
-n 数据库名称不指出的话默认为输入文件名
更多选项请参阅解压后的doc文件夹的formatdb.html文件
进行 BLASTBLAST搜索
在命今行下录入blastall命令及相应的参数
打开输出文件分析结果,如果结果不好可以试着调整参数再次进行BLAST
如下所示命令:
blastall-p blastn-d db_name-i QUERY -o out.QUERY
Blastall 的一些参数说明
-p 程序名包括
blastp: 通过蛋白质序列搜索蛋白质序列数据库
blastn: 通过核酸序列搜索核酸数据库
blastx: 通过翻译后的核酸序列搜索蛋白质数据库
tblastn: 通过蛋白质序列搜索翻译后的核酸数据库
tblastx: 通过翻译后的核酸序列搜索翻译后的核酸数据库
-d 数据库名称与formatdb中-n选项一致
-i 输入文件不指明的话默认为STDIN
-o 输出文件不指明的话默认为STDOUT
-e:设置e-value
-m:比对结果显示格式选项,缺省值为0 ,即pairwise格式。另外还可以根据不同的需要选择1~6等不同的格式。
-I:在描述行中显示gi号[T/F],缺省值F
-b:显示的比对结果的最大数目,缺省值250
-F:对于要查询的序列做低复杂度区域(low complexity regions, LCR)的过滤[T/F],缺省值T。对blastn用的是DUST程序,其他比对用的是SEG程序。
-G: 打开一个gap的罚分(0表示使用缺省设置值),默认0
-E: 扩展一个gap的罚分(0表示使用缺省设置值),默认0
-q: 一个核酸碱基的错配(mismatch)的罚分(只对blastn有效),缺省值-3
-r : 一个核酸碱基的正确匹配(match)的奖分(只对blastn有效),缺省值1
-M: 所使用的打分矩阵,缺省值BLOSUM6244
Bl2seq(两条序列的BLAST)
bl2seq的绝大部分参数是与通用检索程序blastall一致的,只是没有了-d 的选项。另外增加了两个输入选项:
-i:第一个输入序列文件
-j:第二个输入序列文件
注:这两个输入序列都应该是FASTA格式,各自的序列类型–核酸或蛋白–应由所选择的-p 参数决定
命令如下所示:
bl2seq -i query.fa-j sbjct.fa-e 0.01 -o out
Psi Psi-BLAST
Psi-BLAST是由blastpgp命令实现的,它的大部分参数是与blastall一致的,只有少数与迭代检索相关的选项是特别的:
-j: 最大迭代检索的次数,缺省值1,即等同与在blastall中所使用blastp程序
-h: 在每轮检索后构建新的打分矩阵时所选择的序列的期望值(E value)的阈值,缺省值0.001
-C: 将生成的位点特异性的打分矩阵输出到一个文件(二进制格式)
-R: 从文件读取一个原先输出的位点特异性的打分矩阵,然后使用这个矩阵来继续进行以后的检索比对
-Q: 输出一个可读的文本(ASCII)格式的PSI-BLAST的打分矩阵
-B: 设置让blastpgp读取一个已经存在的多重比对文件来构建位点特异性的打分矩阵而进行以后的检索
如下命令所示:
blastpgp-i query.faa-d db_name-o query_out
Fastacmd从数据库中提取序列
-a:是否提取重复accession号的序列[T/F]
-l :设置输出的序列文件每行的字符数
-t :设置在FASTA格式的序列的描述行中只包含gi号[T/F]
-o:输出文件名
命令如下:
fastacmd-d db_name-s p38398
Source from helixnet